What Is SaMD
Software as a Medical Device serves medical purposes without being part of a hardware device. The same AI algorithm can be unregulated wellness software or a high-risk Class III device depending on the clinical claims made.







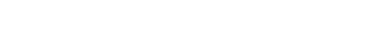
Not all healthcare software is FDA-regulated — the key factor is intended use. Software that analyzes medical images to detect cancer or recommends a diagnosis is SaMD; scheduling or general health apps are not.

Software as a Medical Device serves medical purposes without being part of a hardware device. The same AI algorithm can be unregulated wellness software or a high-risk Class III device depending on the clinical claims made.
The 21st Century Cures Act exempted administrative support, healthy-lifestyle apps, EHRs, and basic lab tests from FDA regulation. Understanding these boundaries is essential before investing in QMS infrastructure.
Class I devices present minimal harm and are subject only to general controls — registration, device listing, and GMP. Most Class I SaMD is exempt from premarket notification.
Class II devices present moderate risk and typically require a 510(k) showing substantial equivalence to a legally marketed predicate. Most first-time SaMD clearances use this pathway.
Class III devices present the highest risk and require premarket approval (PMA) with clinical evidence of safety and effectiveness. Rare for pure SaMD.
Quality by design means regulatory decisions are made before development begins, not retrofitted after. These five steps define it in practice.
A 510(k) is the most common pathway to FDA clearance for Class II SaMD. It must demonstrate substantial equivalence to a legally marketed predicate — same intended use, and technological characteristics that raise no new safety or effectiveness questions.
The submission opens with a precise device description and intended use statement — the same one locked before development, and the foundation the reviewer evaluates everything else against.
The predicate comparison is the core of the 510(k). It must show the same intended use and demonstrate that any differences in technological characteristics don't raise new safety or effectiveness questions.
All SaMD submissions require a Software Description Document and SDLC summary. Documentation depth scales with software safety class — Class C requires the most complete design, testing, and risk management records.
Performance data and FDA-compliant cybersecurity documentation are required for all SaMD. AI diagnostic algorithms also need clinical performance study data against a reference standard.
FDA clearance begins post-market regulatory obligations that continue for the life of the product.

Serious events must be reported to the FDA as Medical Device Reports — within 30 days, or 5 for urgent cases. Complaint handling SOPs are among the first things reviewed during an FDA inspection.
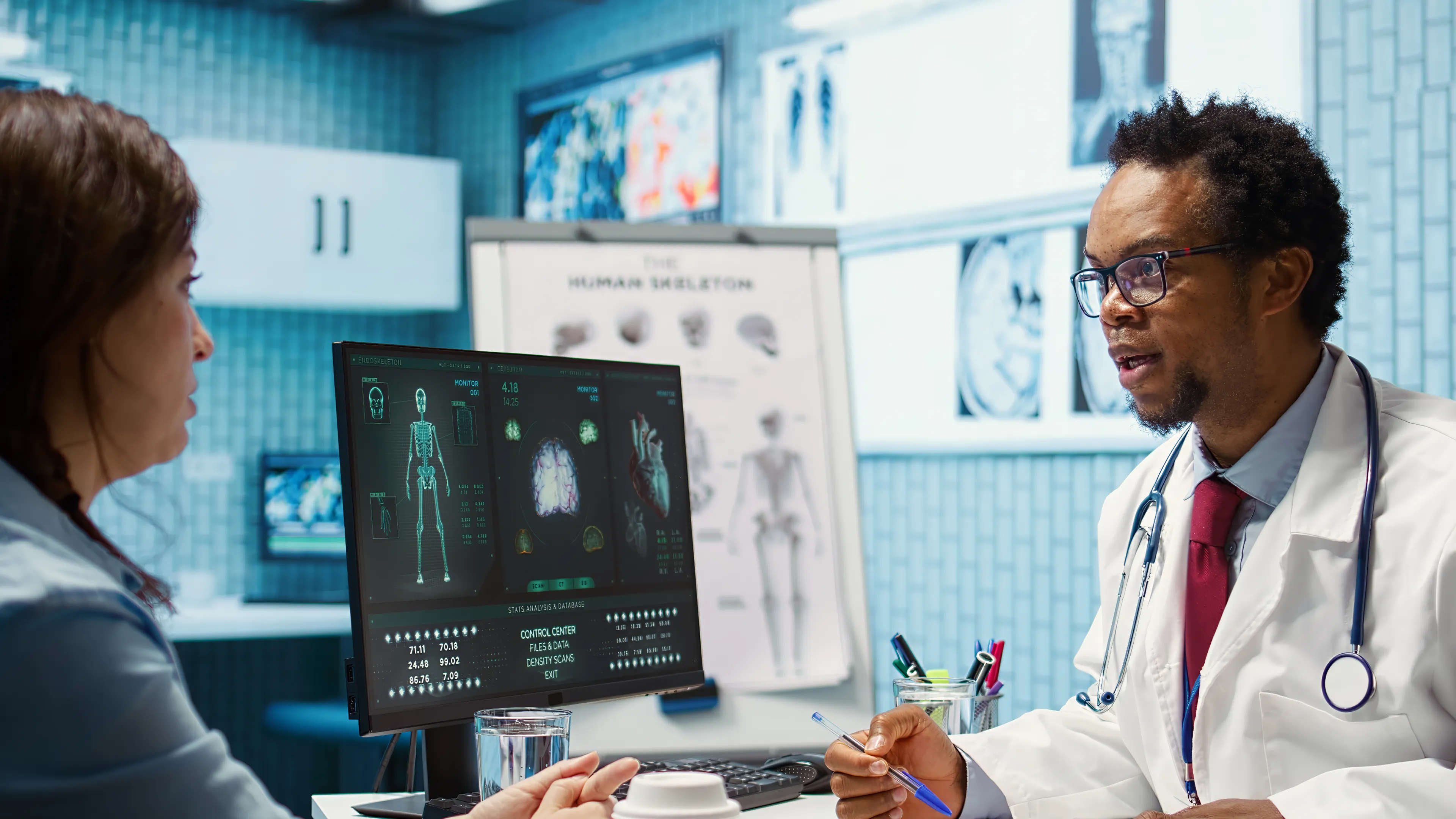
Changes affecting safety, intended use, or labeling may require a new 510(k). Whether a change is significant is itself a regulated decision documented within the QMS.

Annual reports summarizing complaint data, MDRs, and field corrective actions must be submitted to the FDA on schedule — regardless of whether adverse event data exists.

The FDA expects ongoing monitoring of real-world AI/ML SaMD performance against 510(k) claims. Significant degradation must be investigated and reported.

A PCCP lets companies pre-specify AI algorithm modifications that can proceed without a new 510(k), within performance boundaries accepted at clearance.
The most consequential SaMD decisions are made before a line of code is written. These are the five areas where regulatory strategy either prevents problems or creates them.
Book a Regulatory ConsultThe most common and expensive SaMD submission problems are predictable — and preventable with the right regulatory strategy from the start.
Underrating a device's risk class derails a submission mid-review. It takes careful analysis of product codes, predicates, and FDA SaMD guidance.
A backfilled QMS is a top red flag — design control records can't be created after the fact. It must be active before the first design input.
The FDA judges your development process, not just test results. Common gaps: weak traceability, undocumented architecture, and tests not linked to requirements.
FDA rules for SaMD now mandate a management plan, SBOM, threat modeling, and penetration testing evidence. Gaps drive many information requests.
For AI diagnostic algorithms, the clinical performance study underpins your intended use claims. A weak population, reference standard, or analysis plan can trigger a new study.
Complaint handling, MDR reporting, and change control must be live on clearance day. A complaint before the process exists is an immediate compliance problem.
We guide SaMD teams from intended use through 510(k) clearance — QMS setup, predicate selection, clinical validation, and submissions. Our regulatory and engineering teams work together because the documentation and the code are inseparable.
Schedule a Regulatory Call
100 Fastest Growth Companies
Global Spring Winner
Top App Development Company
AWS Partner Network
Google Cloud Partner
Highly Rated on Trustpilot
Verified Agency
Top App Development Company
ASSOCHAM Member
Costs vary with device complexity, clinical study requirements, and QMS maturity. Straightforward SaMD clearances typically run $200,000–$500,000; AI diagnostics requiring prospective validation can reach $1M–$5M or more. Budget for regulatory affairs from day one.
Yes — many digital health products fall under both frameworks. A SaMD handling PHI must meet HIPAA's Security Rule and FDA's QMS and premarket notification requirements. The two create cumulative obligations, so teams need expertise in both domains.
The FDA's predetermined change control plan lets manufacturers pre-specify how an algorithm may evolve and get those modifications accepted at clearance. The framework is still developing — engage the FDA's Digital Health Center of Excellence early.